
- “Studies of bacterial type III secretion systems”
- “Enzymes for DNA modification- biomedical applications”
- “Protein folding”
- “Enzyme structures and mechanisms”
- “Development of methods”
Studies of bacterial type III secretion systems
Type III secretion systems (TTSS) enable plant and animal bacterial pathogens to deliver virulence proteins into the cytosol of eukaryotic host cells causing a broad spectrum of diseases including bacteremia, septicemia, typhoid fever and bubonic plague in mammals and localized lesions, systemic wilting and blights in plants. In addition, type III secretion systems are also required for biogenesis of the bacterial flagellum.
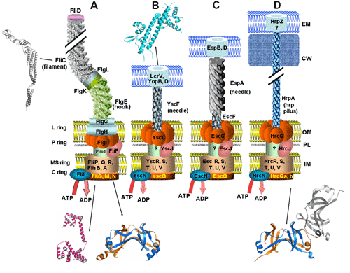
Schematic representation of the bacterial flagellum (A) and the TTSS in Yersinia (B), Eschericia coli (C) and Pseudomonas syringae (D). The basal body of flagellum consists of proteins organizing the C, MS, P and L rings. Only conserved proteins in the TTSS systems (and their flagellar homologues) have drawn and are marked by similar position and coloring. Structurally characterized proteins are shown schematically.
Our group has initiated in collaboration with the group of Prof. Panopoulos (Plant Sciences Section of IMBB) a study of structure-function of TTSS proteins and of their supramolecular assemblies. Work has initially focused on the HrcQB protein, a component of the conserved core of the secretion apparatus of Pseudomonas syringae with homologues in all TTSS. The protein has a variable amino-terminal and a conserved carboxy-terminal domain (HrcQB-C). We have recently determined crystal structure of HrcQB-C (first structure of a conserved TTSS component) and have shown that this domain retains the ability of the full-length protein to interact with other type III components [Fadouloglou et al., PNAS 101, 70-75 (2004)].
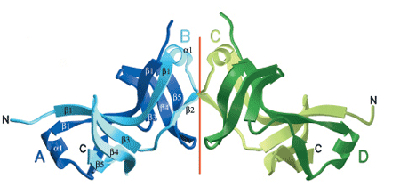
Schematic representation of the HrcQB-C structure. Each chain is individually coloured and labelled.
A 3D-analysis of sequence conservation patterns in HrcQB-C reveals two clusters of residues potentially involved in protein-protein interactions. Based on the analogies between HrcQB and its flagellum homologues, we proposed that HrcQB-C participates in the formation of a C-ring-like assembly. The studies of several other TTSS proteins (conserved core proteins, chaperones and effectors are presently in progress.
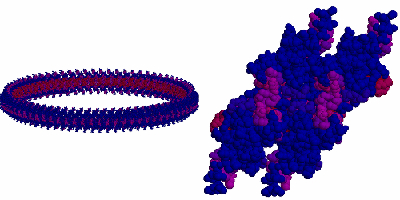
A model for the assembly of HrcQB-C tetramers in a C-ring-like structure based on packing interactions found in the crystal
Enzymes for DNA modification- biomedical applications
The exploitation of the information generated by genomic sequencing efforts in biomedicine and biotechnology will depend (among other things) critically on the availability of DNA binding/modifying enzymes which can target and modify individual genes. Therefore, there is a clear and compelling role for protein crystallography in providing the structural basis for the function of these enzymes. Our group has a 10-year experience in structural studies of DNA binding enzymes (restriction endonucleases, DNA methyltransferases, zinc fingers). Our recent study of PvuII mutants has for the first time shed light on the structural basis of the pathway by which metal ions as essential cofactors enter the catalytic center of restriction endonucleases.
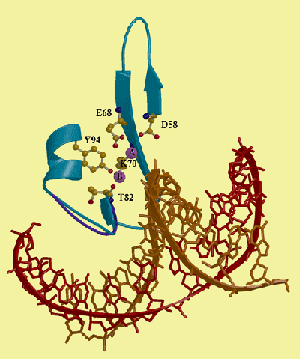
Essential cofactor (Mg2+) binding sites of PvuII restriction endonuclease [Spyridaki et al., JMB 331, 395-406 (2000)
In the framework of a EU project coordinated by us ( http://www.stepc.gr/ENGEM/ ), a major joint effort was undertaken to develop restriction endonucleases and DNA methyltransferases of utmost and programmable specificity for DNA modification and potential biomedical applications (gene replacement, gene silencing). Using restriction endonucleases and DNA methyltransferases as model systems, the project explored three different coupling methods of these proteins to triple-helix-forming oligonucleotide or PNA specificity tags which can make the specificities of the enzyme-oligo/PNA conjugates programmable: 1) classical coupling using heterobifunctional crosslinking reagents, 2) native chemical peptide ligation (NCPL), and 3) intein-mediated protein ligation (IPL). The biochemical, biophysical and structural characterization (via X-ray crystallography) of the interactions with DNA has been carried out. Protocols for the site-specific interactions of the conjugates with DNA have been developed and applications in targeted DNA cleavage and gene silencing are being developed. Our contribution to this project is the development and structural analysis of a novel, monomeric form of the PvuII restriction endonuclease (scPvuII) which provides a convenient system for targeted DNA cleavage and other biomedical applications. Crystallographic and SAXS studies have recently confirmed an unexpected, “semi-open” form of the DNA-free form of this engineered enzyme.

Crystal structure of the engineered scPvuII used for the development of programmable DNA-specificities (the arrow shows the linker introduced to join the subunits of the dimeric wild-type PvuII so as to obtain a monomeric protein). scPvuII has been coupled with triple-helix-forming oligonucleotides and used for targeted DNA cleavage experiments in the framework of the ENGEM project (http://www.stepc.gr/ENGEM/). The long-term goal is to use this protein for a wide range of biomedical applications (e.g. gene replacement).
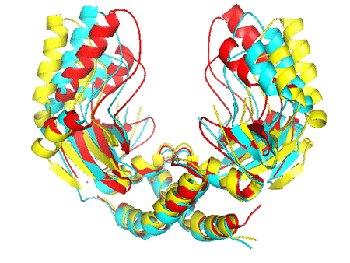
Comparison of the DNA-free form of PvuII (yellow), the scPvuII (blue), and the DNA bound form of PvuII (red) showing the unusual semi-open form of scPvuII in the apo-enzyme form.
Furthermore, we have recently crystallized the DNA-free and DNA-bound forms of M.BseCI DNA methyltransferase, a 67 kDa protein from B. stearothermophilus which methylates the N6 atom of the 3’ adenine in the sequence 5’-ATCGAT-3’.
Protein folding
For several years we have been working on the problem of protein folding using a combination of experimental and computational techniques. The main focus of this work was the development of sequence-structure-stability relationships for the 4-a-helical bundle motif in proteins and the elucidation of factors associated with the establishment of specific topologies in 4-a-helical bundles. The Repressor of primer (Rop) protein, our model system for protein folding studies, is the paradigm of a canonical 4-a-helical bundle. Our group was the first to design a protein mutant (the A31P mutant of Rop) that totally changes its fold (compared to the native structure) as a result of a point mutation; in addition the structural differences between different crystal forms of A31P have been found to be unexpectedly large. Recent molecular dynamics simulations suggest that these structures may correspond to images of two conformers taken from a structurally heterogeneous population of molecules at equilibrium. Based on these findings, we proposed that the A31P may correspond to a kinetically trapped molten globule collaboration with Dr. N.M. Glykos, Univ. of Thrace).

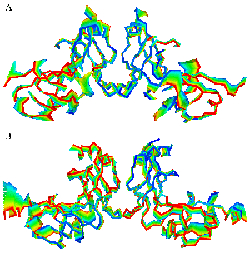
Comparison of wild-type Rop with the structures of mutant A31P in the orthorhombic (A) and the monoclinic (B) crystal forms. The structures of the two crystal forms have been superimposed in C. All structures shown in this figure have been oriented in such a way as to align (both in position and orientation) the helix lying furthest away from the viewer.
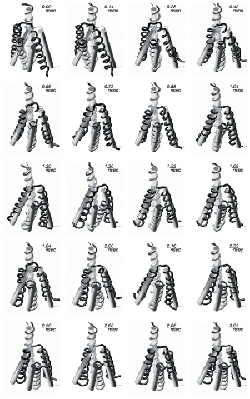
Snapshots recorded from the A31P molecular dynamics trajectory at the indicated times. The cartoon (cylinder) representations correspond to the monoclinic form crystal structure whose orientation and position is kept fixed throughout this figure. The trajectory-recorded structures are depicted as tubes (with different shades of gray used for each monomer) and are oriented in such a way as to align (both in position and orientation) the helix lying furthest away from the viewer with the equivalent helix from the monoclinic form crystal structure. The initial (t=0) trajectory structure is the orthorhombic form A31P crystal structure.
Molecular dynamics simulations have been also used to test the hypothesis that a tetramer analogous to the crystallographically determined HrcQB-C is the building block of a supramolecular structure within P.syringae TTSS resembling the flagellum C-ring structure. Our recent results confirm this hypothesis and strongly suggest a similar building block based on the FliN-C protein (for which also MD simulations were performed) for the flagellum.
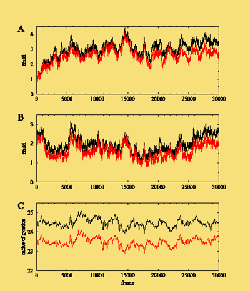
A 12 ns molecular dynamics simulation of the HrcQB-C tetramer performed in explicit water. : (A) rmsd from the initial structure, (B) rmsd from the average structure, and (C) radius of gyration versus frame (each frame equals with 0.4 ps). Black color plots: all the atoms used in calculations. Red color plots: the flexible N-terminal residues are excluded from the calculations.
Principal component analysis indicating that the dominant movement takes place around the dimer-dimer interface. (A) HrcQB-C tetramer, (B) the model of the FliN-C tetramer. The succession of colors indicates the direction of movement.
Enzyme structures and mechanisms
This project deals with the crystallographic elucidation of the 3D-structures of important enzymes and the development of structure-function relationships as a prerequisite for a wide range of applications in biotechnology and health.
We have recently determined the crystal of type I chloramphenicol acetyltransferase (CAT). CAT is responsible for bacterial resistance to chloramphenicol by catalyzing the inactivation of the antibiotic by acetyl group transfer from acetyl CoA to one or both hydroxyl groups of chloramphenicol. Type I CAT (CAT-I) possesses some unique properties which are not observed in other CAT variants. The crystal structure of CAT-I has been determined by our group in the free enzyme form and complexed with fusidic acid. These structures provide information about the catalytic mechanism and the mode of binding to fusidic acid.
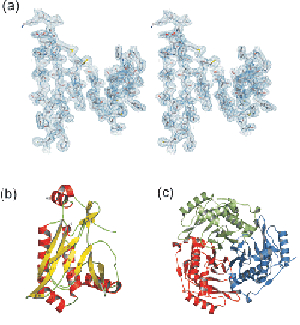
Crystal structure of CAT-I. (a) Detail from the electron density map at 2.5 A, (b) the monomer, (c) the complete (trimeric) enzyme.
In collaboration with the Enzyme Technology and Function group of IMBB (Prof. V. Bouriotis) we study the structural basis of catalytic mechanisms of enzymes from the carbohydrate esterase family 4 (which includes chitin deacetylases, acetyl-xylan esterases, xylanases, rhizobial NodB chitooligosaccharide deacetylases and peptidoglycan deacetylases) and from deacetylases of the LmbE family. We recently crystallized one polysaccharide deacetylase and one LmbE deacetylase from B.cereus. Major enzyme technology applications and design of novel antimicrobials are expected to result in the long term from these studies. The B.cereus enzymes have a particular importance due to their very high homologies to their B.anthracis counterparts; insights into their structures may provide the basis for drugs to counter bioterrorism.

Crystal of one polysaccharide deacetylase (PDA4) obtained in collaboration with the Enzyme Technology & Function group. The protein crystallizes in C222 with a=124.2, b=139.1, c=246.2 A
Development of methods
Several of our projects have required the development of new crystallographic methods. For example, determination of the second conformation of the A31P was only possible after we developed a novel approach to molecular replacement (Multidimensional Molecular Replacement). The software developed by us has been made freely available to the academic community. In the framework of an EU project we worked towards integration of sequence/ structure information sources and the formats and protocols required to access them. Tight integration with a large number of diverse biological information sources was a crucial aspect of the project which was aiming to provide the end user with a single point of uniform access to the vast amounts of information provided by different biological information sources. Methods developed by us include also new approaches in crystallogenesis, treatment of polyacrylamide gels etc.a