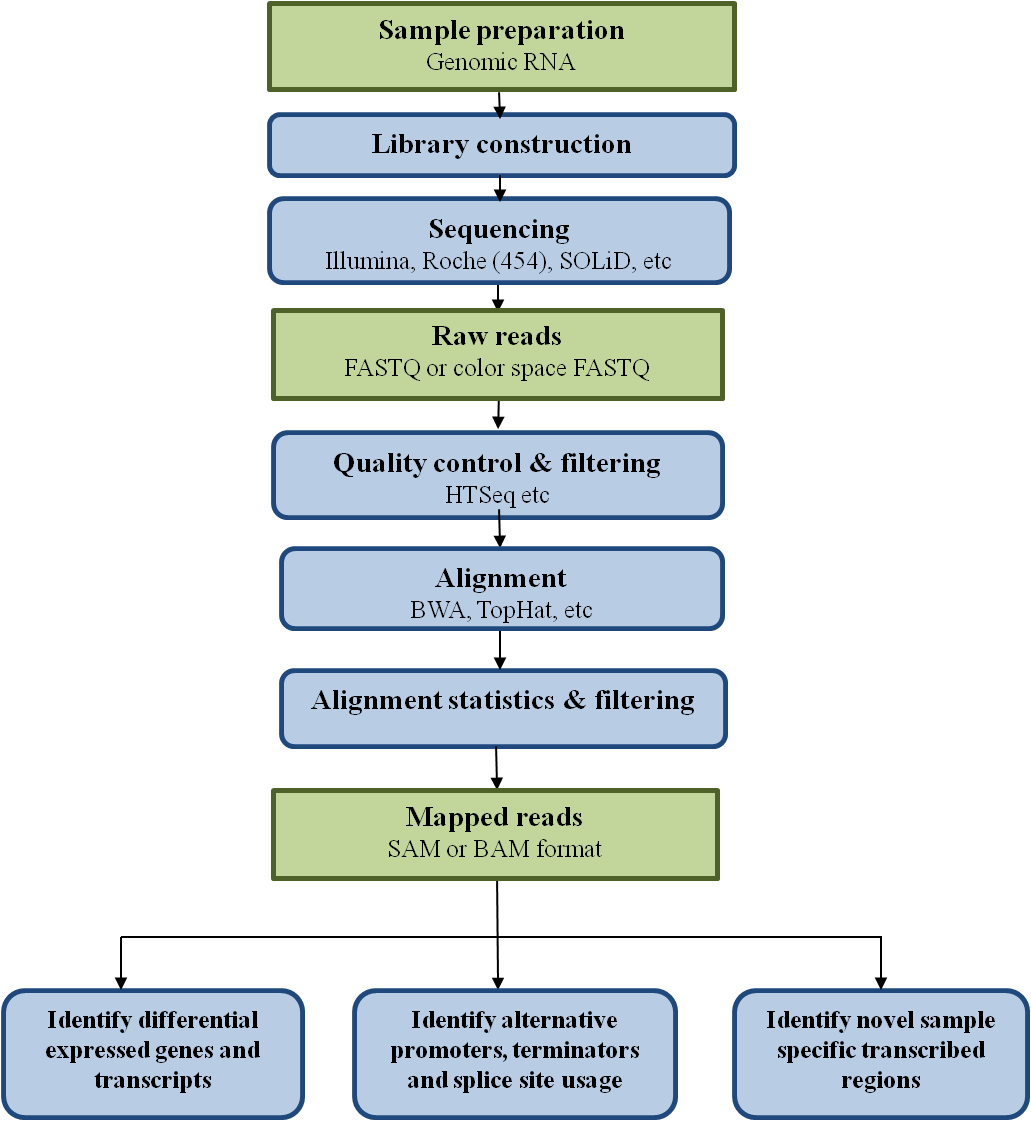
The types of information that can be gained from RNA-seq can be divided into two broad categories: qualitative and quantitative.
- Qualitative data includes identifying expressed transcripts, and identifying exon/intron boundaries, transcriptional start sites (TSS), and poly-A sites. Here, we will refer to this type of information as "annotation".
- Quantitative data includes measuring differences in expression, alternative splicing, alternative TSS, and alternative polyadenylation between two or more treatments or groups. Here we focus specifically on experiments to measure differential gene expression (DGE).
Please keep in mind that the nature of questions people may address using RNA-seq data is effectively limitless, so there are even more facets to the analysis of transcriptomes than there are to generating the data itself. As a disclaimer, we emphasize that it is virtually impossible to stay abreast of all developments in approaches to analyzing RNA-seq data. Our objective rather is to provide in the following outline a rough guide to commonly encountered steps and questions one faces on the path from raw RNA-seq data to biological conclusion.
To generalize, however, embarking on an RNA-seq study usually requires initial filtering of sequencing reads, assembling those reads into transcripts or aligning them to reference sequences, annotating the putative transcripts, quantifying the number of reads per transcript, and statistical comparison of transcript abundance across samples or treatments.